

| 产品编号 | bs-6305R |
| 英文名称 | Ryanodine Receptor Rabbit pAb |
| 中文名称 | 心肌兰尼碱受体抗体(脑肌兰尼碱受体) |
| 别 名 | CCO; CMYO1A; CMYO1B; CMYP1A; CMYP1B; KDS; MHS; MHS1; PPP1R137; RYDR; RYR; RYR-1; SKRR; RYR1_HUMAN; RYR1; Skeletal muscle calcium release channel; Skeletal muscle ryanodine receptor; Skeletal muscle-type ryanodine receptor; Type 1 ryanodine receptor; |
 |
Specific References (1) | bs-6305R has been referenced in 1 publications.
[IF=2.466] Gerburg Keilhoff. et al. The Ryanodine receptor stabilizer S107 fails to support motor neuronal neuritogenesis in vitro. Tissue Cell. 2021 Dec;73:101625 IF ; Mouse.
|
| 研究领域 | 肿瘤 心血管 通道蛋白 细胞膜受体 |
| 抗体来源 | Rabbit |
| 克隆类型 | Polyclonal |
| 克 隆 号 | |
| 交叉反应 | Mouse (predicted: Human,Rat,Rabbit,Pig,Cow,Dog) |
| 产品应用 | IHC-P=1:100-500,IHC-F=1:100-500,IF=1:100-500
not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| 理论分子量 | 566 kDa |
| 细胞定位 | 细胞浆 细胞膜 |
| 性 状 | Liquid |
| 浓 度 | 1mg/ml |
| 免 疫 原 | KLH conjugated synthetic peptide derived from human Ryanodine Receptor: 4701-4800/5038 |
| 亚 型 | IgG |
| 纯化方法 | affinity purified by Protein A |
| 缓 冲 液 | 0.01M TBS (pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol. |
| 保存条件 | Shipped at 4℃. Store at -20℃ for one year. Avoid repeated freeze/thaw cycles. |
| 注意事项 | This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
| PubMed | PubMed |
| 产品介绍 |
The Ryanodine Receptor (RyR) is the channel responsible for calcium release from muscle cell Sarcoplasmic Reticulum (SR) and also plays a role in calcium regulation in non-muscle cells. The RyR exists as a homotetramer and is predicted to have a short cytoplasmic C-terminus and 4-10 transmembrane domains. The remainder of the protein, termed the "foot" region, is located in the cytoplasm between the transverse tubule and the SR. Mammalian RyR isoforms are the product of three different genes: RyR-1 is expressed predominantly in skeletal muscle and areas of the brain; RyR-2 is expressed predominantly in heart muscle but also found in the stomach, endothelial cells and diffuse areas of the brain; and RyR-3 is found in smooth muscle and the brain (striatum, thalamus and hippocampus). In non-mammalian vertebrates, the RyR isoforms are termed alpha, beta and cardiac which correlate loosely to the mammalian RyR-1, RyR-3 and RyR-2 isoforms respectively. Function: Calcium channel that mediates the release of Ca(2+) fromthe sarcoplasmic reticulum into the cytoplasm and thereby plays akey role in triggering muscle contraction following depolarizationof T-tubules. Repeated very high-level exercise increases the openprobability of the channel and leads to Ca(2+) leaking into thecytoplasm. Can also mediate the release of Ca(2+) fromintracellular stores in neurons, and may thereby promote prolongedCa(2+) signaling in the brain. Required for normal embryonicdevelopment of muscle fibers and skeletal muscle. Required fornormal heart morphogenesis, skin development and ossificationduring embryogenesis (By similarity). Subunit: Homotetramer. Can also form heterotetramers with RYR2.Interacts with CALM; CALM with bound calcium inhibits the RYR1channel activity. Interacts with S100A1. Interacts with FKBP1A;this stabilizes the closed conformation of the channel. Interactswith CACNA1S; interaction with CACNA1S is important for activationof the RYR1 channel. Interacts with CACNB1. Interacts with TRDN andASPH; these interactions stimulate RYR1 channel activity (Bysimilarity). Identified in a complex composed of RYR1, PDE4D, PKA,FKBP1A and protein phosphatase 1 (PP1). Repeated very high-levelexercise decreases interaction with PDE4D and protein phosphatase 1(PP1). Subcellular Location: Sarcoplasmic reticulum membrane; Multi-pass membrane protein (Probable). Membrane; Multi-pass membrane protein. Microsome membrane; Multi-pass membrane protein. Tissue Specificity: Brain, skeletal muscle, placenta and possibly liver and kidney. In brain, highest levels are found in the cerebellum, hippocampus, caudate nucleus and amygdala, with lower levels in the corpus callosum, substantia nigra and thalamus. Post-translational modifications: Channel activity is modulated by phosphorylation.Phosphorylation at Ser-2843 may increase channel activity. Repeatedvery high-level exercise increases phosphorylation at Ser-2843.[PTM] Activated by reversible S-nitrosylation. Repeated veryhigh-level exercise increases S-nitrosylation. DISEASE: Malignant hyperthermia 1 (MHS1) [MIM:145600]: Autosomaldominant pharmacogenetic disorder of skeletal muscle and is one ofthe main causes of death due to anesthesia. In susceptible people,an MH episode can be triggered by all commonly used inhalationalanesthetics such as halothane and by depolarizing muscle relaxantssuch as succinylcholine. The clinical features of the myopathy arehyperthermia, accelerated muscle metabolism, contractures,metabolic acidosis, tachycardia and death, if not treated with thepostsynaptic muscle relaxant, dantrolene. Susceptibility to MH canbe determined with the 'in vitro' contracture test (IVCT):observing the magnitude of contractures induced in strips of muscletissue by caffeine alone and halothane alone. Patients with normalresponse are MH normal (MHN), those with abnormal response tocaffeine alone or halothane alone are MH equivocal (MHE(C) andMHE(H) respectively). Note=The disease is caused by mutationsaffecting the gene represented in this entry. Central core disease of muscle (CCD) [MIM:117000]:Autosomal dominant congenital myopathy, but a severe autosomalrecessive form also exists. Both clinical and histologicalvariability is observed. Affected individuals typically displayhypotonia and proximal muscle weakness in infancy, leading to thedelay of motor milestones. The clinical course of the disorder isusually slow or nonprogressive in adulthood, and the severity ofthe symptoms may vary from normal to significant muscle weakness.Microscopic examination of CCD-affected skeletal muscle reveals apredominance of type I fibers containing amorphous-looking areas(cores) that do not stain with oxidative and phosphorylasehistochemical techniques. Note=The disease is caused by mutationsaffecting the gene represented in this entry. Multiminicore disease with external ophthalmoplegia(MMDO) [MIM:255320]: Clinically heterogeneous neuromusculardisorder. General features include neonatal hypotonia, delayedmotor development, and generalized muscle weakness and amyotrophy,which may progress slowly or remain stable. Muscle biopsy showsmultiple, poorly circumscribed, short areas of sarcomeredisorganization and mitochondria depletion (areas termed minicores)in most muscle fibers. Typically, no dystrophic signs, such asmuscle fiber necrosis or regeneration or significant endomysialfibrosis, are present in multiminicore disease. Note=The disease iscaused by mutations affecting the gene represented in this entry. Congenital myopathy with fiber-type disproportion (CFTD)[MIM:255310]: Genetically heterogeneous disorder in which there isrelative hypotrophy of type 1 muscle fibers compared to type 2fibers on skeletal muscle biopsy. However, these findings are notspecific and can be found in many different myopathic andneuropathic conditions. Note=The disease is caused by mutationsaffecting the gene represented in this entry. Note=Defects in RYR1 may be a cause of Samaritanmyopathy, a congenital myopathy with benign course. Patientsdisplay severe hypotonia and respiratory distress at birth. Unlikeother congenital myopathies, the health status constantly improvesand patients are minimally affected at adulthood. Similarity: Belongs to the ryanodine receptor (TC 1.A.3.1) family. RYR3 subfamily. Contains 3 B30.2/SPRY domains. Contains 5 MIR domains. SWISS: P21817 Gene ID: 6261 Database links: UniProtKB/Swiss-Prot: P21817.3 Defects in the RYR2 gene are the cause of several heart diseases: 1) familial arrhythmogenic right ventricular dysplasia 2 (ARVD2), also known as arrhythmogenic right ventricular cardiomyopathy 2 (ARVC2), 2) an autosomal dominant form of stress-induced polymorphic ventricular tachycardia (VTSIP), also known as catecholaminergic polymorphic ventricular tachycardia (CPVT) and 3) familial polymorphic ventricular tachycardia (FPVT). Ryanodine Receptor 2 contains several phosphorylatable sites. Specifically, Ser-2030 and Ser-2809 (or at Ser-2808 depending on the species) can be phosphoryated by protein kinase A (PKA) and Ser-2815 (or at Ser-2814 depending on the species) can be phosphorylated by CaMKII (Ca2+/calmodulin-dependent protein kinase II). |
| 产品图片 |
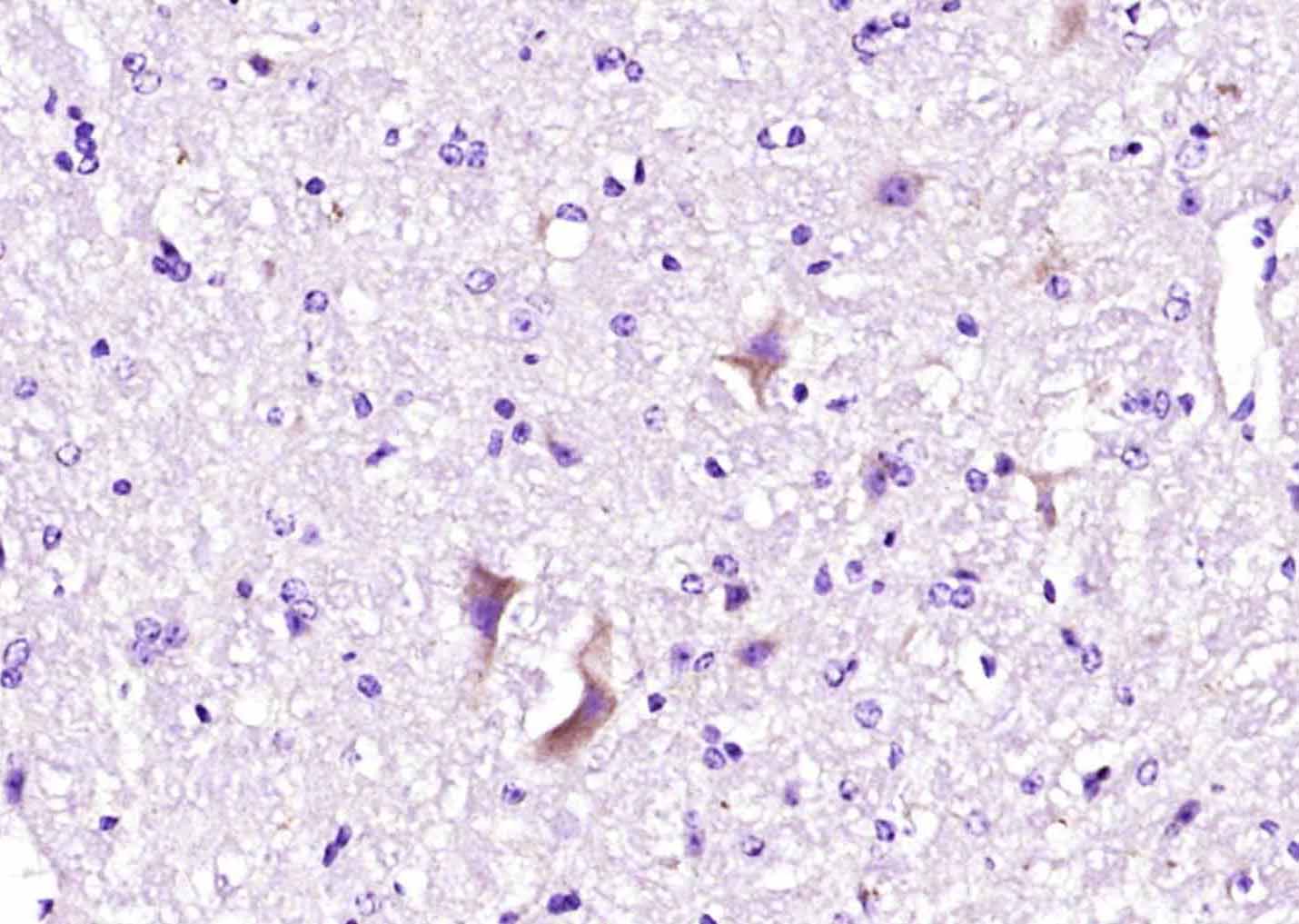
Paraformaldehyde-fixed, paraffin embedded (mouse cerebellum tissue); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (Ryanodine Receptor) Polyclonal Antibody, Unconjugated (bs-6305R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.
|
| 1、抗体溶解方法 | |
| 2、抗体修复方式 | |
| 3、常用试剂的配制 | |
| 4、免疫组化操作步骤 | |
| 5、免疫组化问题解答 | |
| 6、Western Blotting 操作步骤 | |
| 7、Western Blotting 问题解答 | |
| 8、关于肽链的设计 | |
| 9、多肽的溶解与保存 | |
| 10、酶标抗体效价测定程序 | |





